蛋白质是所有细胞过程的关键,其结构对于了解其功能和进化非常重要。基于序列的蛋白质结构预测的准确性不断提高,AlphaFold数据库[1]中已有超过2.14亿个预测结构。然而,在这种规模上研究蛋白质结构需要高效的方法。这里,我们开发了一种基于结构对齐的聚类算法——Foldseek聚类,它可以聚类数以亿计的结构。利用这种方法,我们对AlphaFold数据库中的所有结构进行了聚类,识别出230万个非单例结构簇,其中31%的结构缺乏标注,代表着以前可能未被描述过的结构。未被标注的簇往往只有很少的代表,仅占AlphaFold数据库中所有蛋白质的4%。进化分析表明,大多数集群起源古老,但有4%似乎具有物种特异性,代表了质量较低的预测或新生基因样本。我们还展示了如何利用结构比较来预测结构域家族及其关系,确定远程结构相似性的例子。在这些分析的基础上,我们确定了人类免疫相关蛋白与原核生物物种中推测的远缘同源性的几个例子,说明了这一资源在研究整个生命树的蛋白质功能和进化方面的价值。
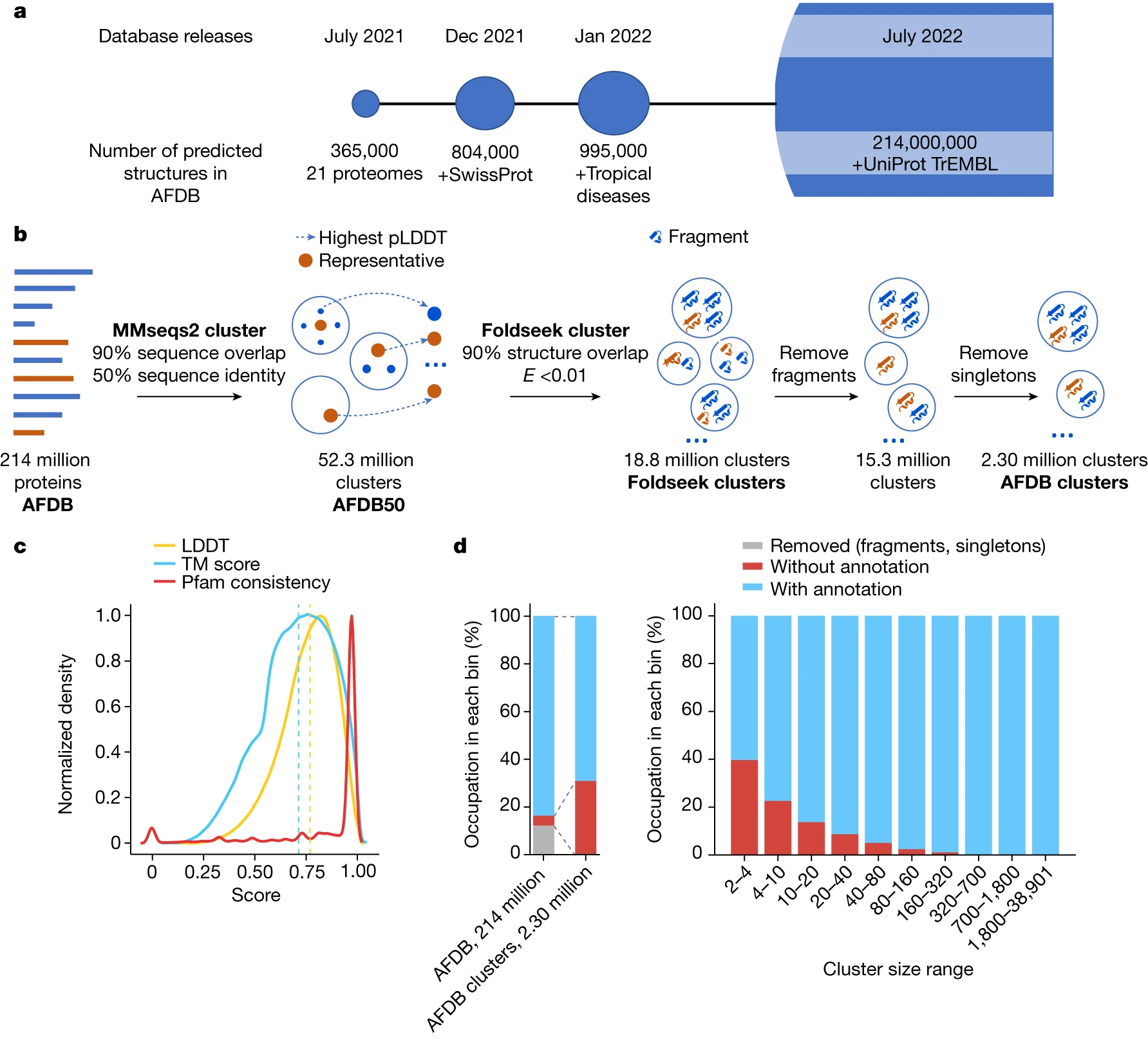
图 1:AFDB、结构聚类工作流程和聚类摘要。
a,AFDB始于2021年,是EMBL-EBI[2]和DeepMind的合作成果。该数据库的发展经历了多个阶段,2022年的最新版本包含了超过2.14亿个预测的蛋白质结构及其置信度指标。
b,采用两步法对数据库中的蛋白质进行聚类。首先,根据50%的序列同一性和90%的序列重叠性,用MMseqs2[3]对2.14亿个UniProtKB蛋白序列(AFDB)进行聚类,从而将数据库规模减少到5200万个聚类(AFDB50)。每个聚类选择pLDDT[4]得分最高的蛋白质作为代表。接下来,使用Foldseek[5]将代表性结构聚类为1,880万个聚类(Foldseek聚类),不设序列同一性阈值,但仍强制要求每个结构比对有90%的序列重叠且E值小于0.01。最后一步,从聚类中剔除了所有标记为片段的序列,最终得到230万个至少有两种结构的聚类(AFDB 聚类)。
c,AFDB聚类结构和Pfam[6]一致性。所有聚类的LDDT中位数为0.77,TM中位数为0.71,66.5%的聚类与Pfam注释100%一致。
d,有注释和无注释的序列和聚类的摘要(左)以及聚类大小与注释的关系(右)。从左到右,每个分区占据AFDB聚类的比率分别为12.24%、10.59%、9.20%、10.07%、10.46%、10.05%、9.04%、9.20%、9.19%和9.96%。
注:
[1] AlphaFold Database,AFDB,即AlphaFold Structure Database,蛋白结构数据库,是一个开放获取的、包含大量高精度蛋白质结构预测数据的数据库。它使得已知蛋白质序列空间结构覆盖率达到了前所未有的扩张。AlphaFold数据库提供了对预测原子坐标、每个残基和成对模型置信度估计以及预测的对齐误差的程序化访问和交互式可视化的功能。AlphaFold是DeepMind公司开发的一个人工智能系统,可以根据蛋白质的氨基酸序列预测蛋白质的结构。AlphaFold前所未有的准确性和速度使得大规模结构预测的数据库得以建立。这将使生物学家能够获得几乎任何蛋白质序列的结构模型,这改变了他们解决研究问题的方式,并加速了他们的项目进展。关于AlphaFold数据库的信息可参考《AlphaFold蛋白质结构数据库的简介》和AlphaFold网站或https://www.alphafold.ebi.ac.uk/。
[2] EMBL-EBI,EMBL(European Molecular Biology Laboratory)是指欧洲分子生物学实验室,总部位于德国海德堡,1974年由欧洲14个国家加上亚洲的以色列共同发起建立,现在由欧洲30个成员国政府支持组成,目的在于促进欧洲国家之间的合作来发展分子生物学的基础研究和改进仪器设备、教育工作等。EBI(European Bioinformatics Institute)是指欧洲生物信息学研究所,是非盈利性学术组织EMBL的一部分,1982年建立了先进的核苷酸序列数据库(EMBL-DNA),可进行核苷酸序列检索及序列相似性查询。综合起来的EMBL-EBI数据库功能是一款针对人类蛋白GO功能分析的综合注释数据库。更多介绍可参考《EMBL-EBI QuickGO:蛋白功能分析数据库》。
[3] MMseqs2,是由Martin Steinegger和Johannes S?ding开发的蛋白质序列搜索软件,声称将PSI-BLAST的速度提升了400倍。更多介绍可参考《使用MMseq2对序列聚类》和《传统蛋白质序列比对算法》。
[4] pLDDT,是是在 0 -100范围内对局部置信度的每个残基的度量。pLDDT可以沿着一条链显著变化,使得模型能够表达结构域的高置信度,但是在结构域之间的连接子(linker)上具有低置信度。研究人员提出了一些证据,证明低 pLDDT 的区域可能是孤立的非结构。pLDDT<50 的区域不应被解释,或者被解释为「可能的无序预测」。更多介绍可参考《高效预测几乎所有人类蛋白质结构,AlphaFold再登Nature,数据库全部免费开放》和《蛋白质构象评价指标》。
[5] Foldseek,是德国马克斯·普朗克多学科科学研究所的Johannes S?ding团队开发了一种蛋白质结构数据搜索方法。随着结构预测方法产生数以百万计的公开可用的蛋白质结构,搜索这些蛋白数据正成为一个瓶颈。作者开发的Foldseek方法通过将蛋白质内的三级氨基酸相互作用描述整理为结构字母表的序列,将查询到的蛋白质结构与数据库进行比对,将计算时间减少了4到5个数量级。更多介绍可参考《Nat Biotech | AI实现快速精准地搜索蛋白质结构》和《蛋白结构生物信息学的造极:AlphaFold DB的结构挖掘》。
[6] Pfam(the protein family database, MSA)收集了大量使用多重序列比对和隐马尔可夫模型(hidden markov models, HMMs)对UniProKB的蛋白质序列数据进行结构域归类形成的蛋白质家族,广泛用于通过序列比对推测蛋白质的结构域排布形式及功能。更多介绍可参考《Pfam蛋白质结构域家族数据库》和《pfam基本介绍,以及蛋白质序列下载》以及《基于Pfam中hmm结构的基因家族分析》。
BMEN202311040003
来源:Nature
标题:Clustering predicted structures at the scale of the known protein universe

